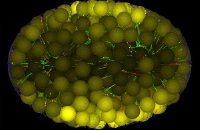
This page describes an always present but usually invisible feature of the skeledyne computer program we use for simulating cytoskeletal dynamics. The skeledyne program is available here.
We use LaGrangian “cytoplasmic domains” to keep track of the diffusion and convection of soluble factors throughout a cell's cytoplasm in a way designed to finesse the famously difficult fluid dynamics free boundary problem. When a deformable boundary (e.g. cell cortex) encloses an incompressible fluid (e.g. cytoplasm), solving the equations of fluid motion requires knowing stress or flux boundary conditions everywhere on the free boundary. But the location of that moving boundary is an unknown to be computed. To compute how the boundary moves in order to know where it is and thus know where to apply boundary conditions on the fluid, one must know what forces the interior fluid exerts on this boundary. But what forces the contained fluid exerts requires knowing how it is moving, i.e. knowing the solution to its equations of motion. This circular coupling between the equations of motion of the contained fluid and the deformable cortex makes this one of the most difficult in fluid dynamics even when the 'fluid' is a featureless linearly viscous fluid (like water or honey). But the cytoplasm in embryonic blastomeres is, instead, a dense soup of various sized yolk particles, organelles, and other particulate inclusions, which occupy a large volume fraction, e.g. 50%. And these particles are large relative to the microscale cellular features our work focuses on: microtubules, centrosomes, chromosomes, etc. Such cytoplasm is therefore not well represented by standard viscous fluid models. We therefore use agent-based modeling to represent the cytoplasm as consisting of thousands of nearly close-packed particles of arbitrary size which collide with their neighbors, which fend each other off (i.e. cannot encroach into the volume occupied by other particles), and which exert viscous drag forces on each other as they move relative to each other. Whatever volume those "interesting" particles do not fill up is filled up by spherical "cytoplasmic domains", all of the same diameter. Thus, our cytoplasm is a space-filling mass of particles of various kinds, which all fend each other off and slide viscously past each other. The cytoplasmic domains among them do more than simply filling the space that other organelles do not fill; they play an essential role as biochemical reaction vessels. Each cytoplasmic domain carries a vector of concentrations of soluble proteins with it as it moves, thus modeling convection. These concentrations represent, for examples, mRNAs being translated into proteins, building block monomers or dimers such as GDP-tubulin and GTP-tubulin dimers from which microtubules polymerize, kinesin motors not yet bound to microtubules, etc. To account for biochemical reactions in each cytoplasmic domain, such as mRNAs being translated into proteins, GDP-tubulin being converted to GTP-tubulin, phosphorylation/dephosphorylation of proteins, degradation of factors, etc. we use mass action kinetic equations (this saves computer time compared to representing each soluble molecule stochastically and individually). Soluble factors in cytoplasmic domains exchange between neighboring domains, thus modeling diffusion in a standard way. Eventually we will install a full version of our Ingeneue genetic network simulator code inside each cytoplasmic domain. Each particle representing a nucleus is also a cytoplasmic domain, and only these nuclear domains participate in transcription of mRNAs, which they export diffusively into neighboring cytoplasmic domains. Yolk particles exclude all gene products and thus form flux-impeding obstacles. In our model, all spherical particles collide and fend each other off, whereas microtubules and other thin filaments pierce through cytoplasmic domains exchanging only viscous drag forces, not collision forces. To facilitate efficient calculation of diffusive fluxes among cytoplasmic, each cytoplasmic domain agent keeps a list of pointers to its neighboring domains. Domains that move past each other, or past the organelles mentioned above, or past microtubules, exchange fluid viscous drag forces. This exchange of viscous drag forces, together with exchange of forces that prevent domains from encroaching into each others space, make the ensemble of cytoplasmic domains and organelles act like an incompressible viscous fluid, and the implementation of mass action kinetic equations in each domain, and diffusive flux equations between adjacent domains, is an agent-based method for solving reaction-diffusion-convection conservation equations in that viscous fluid.
Our modeling of microtubule dynamics illustrates the interaction between agents and cytoplasmic domains. By keeping track of the locations of a myriad close-packed cytoplasmic domains, and the numbers of soluble factor molecules in each one, we have a way to determine the concentration of any soluble factor near any 3-D point, x, in the cell; we take it to be the concentration in the cytoplasmic domain nearest to x. Thus, for example, when x is the current location of a polymerizing MT tip, we can compute the concentration of GTP-tubulin near x, and thus compute how fast that tip should elongate (until it hits some obstacle). When MTs depolymerize, they return the tubulin dimers that formed them to the nearest cytoplasmic domains. This results in spatio-temporal gradients of different forms of tubulin dimer, and the resulting competition for dimers has a strong effect on MT dynamics in large cells (such as 160 um diameter green urchin zygotes or 50 um long worm embryos). This jumping of tubulin dimers between soluble pools in cytoplasmic domains and specific parts of a MT agent, which hydrolyzes its own GTP according to stochastic kinetics, is just one instance of exchange between continuous (soluble) and individual agent forms.
Another example is kinesin motors. At any given moment, those motors not bound to MTs are dispersed in soluble form among all the cytoplasmic domains. For each MT segment, we can compute the concentration of soluble kinesin nearby, and compute from the probability (during each time step) that a kinesin will bind to that MT segment (becoming a newly created individual agent that will walk along the MT and depleting, abruptly, the MKLP1 concentration in the cytoplasmic domain it came from). When, later on, that MKLP1 falls off its MT, it will disappears as individual agent, and boost, abruptly, the concentration of soluble MKLP1 in the nearest cytoplasmic domain.
Using cytoplasmic domains interspersed among other solid particles like yolk particles, with a mass-action kinetic simulation instantiated separately in each one, would be little different from using a standard fluid dynamics Eulerian mesh discretization of reaction and diffusion in the fluid until the issue arises of meeting boundary conditions (for the fluid phase) on the surface of myriad suspended solid particles, each of which moves in response to forces the fluid exerts on it. The embedded 'solid' particles make the fixed Eulerian mesh approach too computationally expensive and gives the advantage to using our swarm of organelles and cytoplasmic domains (which are analagous to classic LaGrangian-coordinate finite elements, and which we represent as individual agents). Our cytoplasmic domains have another advantage which is important to our proposed future work: A swarm of mostly spherical particles which are incompressible in the sense that they fend each other off, can be compressed by a contractile cell cortex only so far, after which the inward forces, which the cortex exerts on the particles it contains, come into balance with the repulsive forces that prevent particles from encroaching into neighboring particles. Balancing inward cortical forces with outward particle collision forces, in effect, solves the moving free boundary problem using our collision detector, as follows.
Since the number of MT segments and MKLP1 motors (in the tens of thousands) greatly exceeds the number of cytoplasmic domains we require (about one thousand), the extra time required to detect cytoplasmic domain collisions is an insignificant increment. Also the number of differential equations (3 per domain) needed to keep track of the position of each domain is a small fraction of all the ODEs in the simulation. This means that the computational infrastructure already in our model can compute the trajectories of all the cytoplasmic domains for a small additional computational cost.
The most computationally difficult part of our agent-based 3D cytoskeletal dynamics simulator is detecting collisions between parts as they move around. Any part can move anywhere. We have devised a scheme for detecting their collisions in which the computer time required increases in linear proportion to the number of parts involved, even though the number of possible collisions rises quadratically with the number of parts. Moreover, we have written threaded code so that, on the multiple processor computers we use, one thread running on one processor can detect collisions while other threads running on other processors can compute the consequences of those collisions. Among collisions our scheme detects are those between the generic cytoplasmic domain spheres and other "real" agents. We must detect those collisions anyway in order to know, for example, which cytoplasmic domains are nearest MT tips that take tubulin dimers from them as they polymerize. Detecting those collisions, necessary for other purposes, leads also to automatic generation of the repulsive forces between particles that makes the swarm of space-filling particles behave as an incompressible deformable mass. So far, our model cell cortex cannot deform or contract, but will in future, and we already have the LaGrangian coordinate finite elements in place, moving, carrying out interior biochemical reactions plus diffusive exchange with their neighboring domains, and acting as an incompressible mass in preparation to resist unchecked contraction by the future contractile cortex. This will lead to simulations in which a cell determines its own 3-D boundaries, in whatever shape achieves a force balance between inward forces the cortex generates to minimize its area and outward forces the particles filling the interior generate to fend off each other and the cortex.
Below are other sample simulations, made by the skeledyne simulation program (available here), in which cytoplasmic domains play a crucial role.
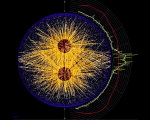
| 140.7 MB [675 x 640 pixels] |
MKLP1 motors on microtubules in urchin egg(Garrett Odell & Victoria Foe)